前言
由于正在搞毕设,需要做结构优化(与优化相对的是单点计算 所谓单点计算 就是不优化结构 直接算能量 电子相关的性质:几何结构在计算前后不发生变化)自然第一个想到的就是比较流行的商业软件VASP,用过了几次,虽然结果不太好(多半是我自己模型的问题),但还是写在这里记录一下。最重要的就是不懂要查看手册官网手册(一定要多看手册!多看手册!多看手册!) 。
主要参考 大师兄!learn vasp the hard way
正文
VASP的主要输入、输出文件有四个:
INCAR、POSCAR、POTCAR、KPOINTS、OUTCAR、CONTCAR、OSZICAR(由于仅涉及结构优化,不考虑能带、电子态密度等的计算,暂时先说这些)
INCAR文件
文件中输入一些控制整个计算过程的参数,
SYSTEM
-
SYSTEM 是对整个计算的注释、说明,后面可以随便写,但是尽量不要写中文。
-
SYSTEM=Ar Structure Optimization #注释
EDIFF EDIFFG NSW NELM NELMIN POINT IBRION ISIF
控制电子步和离子步收敛的参数。
先解释一下电子步和离子步的概念: 一般来讲,电子步就是电子自洽迭代的次数(默认60),离子步是离子驰豫的步数。
结构优化也叫结构迟豫。是指对整个输入体系的坐标进行调整,得到一个相对稳定的基态结构。结构优化分原子迟豫和电子迭代两个嵌套的过程,每次计算中都进行原子迟豫和电子迭代计算(电子迭代嵌套在原子迟豫中),达到原子迟豫收敛标准时进行下一步计算,直到达到自动中断或者最大原子迟豫步数。(通常以前后两次总自由能之差或 原子所受最大的力 作为原子迟豫的收敛标准,电子自洽迭代计算以总能作为收敛标准,默认为10-4,因电子迭代的嵌套特性,以及电子迭代和原子迟豫的收敛速度不同,最终以原子迟豫最大步数作为强制停止参数)
-
EDIFFG:原子迟豫收敛标准,力作为收敛标准:EDIFFG为负, 一般-0.01到-0.05之间;能量作为标准:EDIFFG 为正, 一般0.001-0.0001. (-0.01对于力收敛来说已经是一个很严格的要求了)。
-
EDIFF:电子迭代收敛标准 1E-4或者1E-5即可
-
NSE:离子步的最大步数 NSW
-
NELM:电子步自洽迭代最大步数,默认为60
-
NELMIN:最小的电子步迭代次数,默认2
-
POINT:IBRION=1,2,3时表示原子每步移动的大小,默认为0.5。即是使用准牛顿法、共轭梯度法、最速下降法优化原子位置时需要设置的步长
-
IBRION:离子驰豫方式,可以去3(初始结构很差),2(CG方法优化,较慢,找到全剧最小的可能性大),1(准牛顿方法 速度快 适合结构与标准模型接近),-1(原子不移动,是IBRION的默认值,NSW=0是原子也不会移动,静态自洽的时候使用)[IBRION]https://cms.mpi.univie.ac.at/vasp/vasp/IBRION_tag_NFREE_tag.html)
IBRION=2时的计算步骤:
第一步,从初始结构出发,计算体系中离子间的作用力,
第二步,VASP尝试着把离子沿着前面估算的方向移动,尝试移动的大小由POTIM这一项决定,
第三步,计算尝试移动后能量和力的大小,据此加入一个矫正项来控制真实移动的大小;
第四部, 移动后,重新计算能量和力,重复前三步直至能量或者力收敛到我们设置的EDIFFG值。
IBRION = 2 时,对POTIM的依赖性很强,因此我们计算的时候要设置一个合理值。
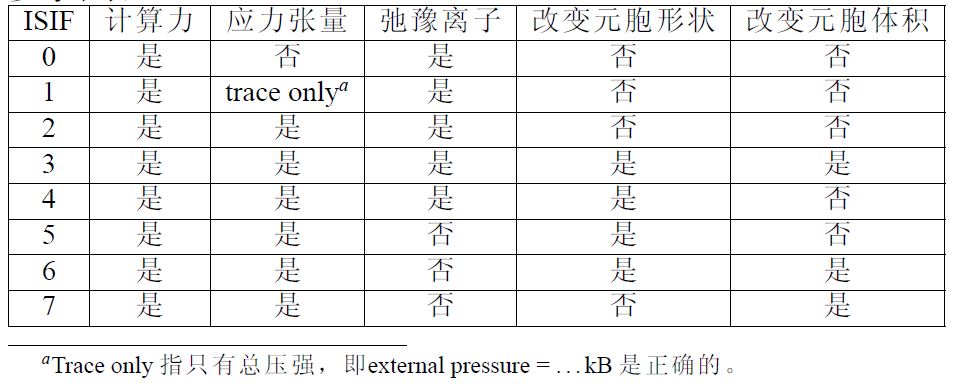
- ISIF 控制原子迟豫过程中晶胞的变化情况 IBRION=0时默认0,其他情况默认2,常用2,3 具体如图:
ISMEAR SIGMA PREC
-
ISMEAR 每个波函数的部分占有数 (还不是很理解)金属使用1,2,半导体使用-5,结构优化不能用-5
-
SIGMA(展开的宽度)的话选定某一个不是很夸张的值应该都可以,如果对于大体系(几百个原子那种)可以选的大一些,到0.1或者再大一些,要是小胞的话可以0.05或者0.01之类的。
-
SIGMA的取值和ISMEAR息息相关
-
ISMEAR = -5 (对于所有体系),SIGMA的值可以忽略,也可以不管(VASP会自动略过)
-
SIGMA的取值和KPOINTS密切相关,Kpoints确定之后,测试SIGMA取值合理性。标准是: grep ‘entropy T’ OUTCAR 得出的能量除以体系中原子的数目,小于0.001 eV 合格
- 对于分子,原子体系(也就是你把分子或者原子放到一个box里面),K点只有一个Γ点取ISMEAR=0,SIGMA必须要用很小的值,如0.01
- MP方法(ISMEAR=1..N):SIGMA取值太大,计算出来的能量可能不正确;SIGMA取值越小,计算越精确,需要的时间也就越多
从经验上来说:对于金属体系,使用MP方法(ISMEAR=1..N)时,SIGMA= 0.10 足够了,官网给的参考值是0.20。
对于高斯展宽Gaussian Smearing (ISMEAR = 0), 对于大部分的体系都能得到理想的结果
SIGMA取值比较大的时候会得到与MP方法相近的误差;但是误差多大,GS方法不可以得到,而MP方法可以。从这一点上来说,MP要比GS好些;
使用GS方法的时候(ISMEAR=0),SIGMA的数值要测试下,保证grep “entropy T” OUTCAR tail -1结果平均到每个原子上小于0.001 eV也就是1meV。不想测试,对于金属体系:SIGMA=0.05是一个很安全的选择
对于半导体和绝缘体,SIGMA取值要小,SIGMA = 0.01 – 0.05 之间也是很安全的。
- PREC 计算精度

ISTART ICHARG INIWAV
-
ISTART 初始波函数产生方法
默认存在WAVECAR时取1,否则0
ISTART=0,根据INIWAV决定初始波函数的产生方法
ISTART=1,波函数从WAVECAR文件读入
-
INIWAV初始波函数产生方法
仅在ISTARTT=0生效
0 凝胶波函数 1[默认]随机数
-
ICHAGE初始电荷密度产生方法
默认ISTART=0取2否则取0
0从初始波函数计算
1从CHGCAR(上次输出的电荷密度)读入
+10非自洽运算,电荷密度在计算过程中保持不变
如1+10=11时,电荷密度保持CHGCAR中的值不变,适用于给定电荷密度求能级本征值(计算能带,输出EIGENVAL文件)和态密度(DOS,输出DOSCAR文件),用于能带计算
ISPIN MAGMOM
由体系决定
- ISPIN=0(默认) 不考虑自旋
- ISPIN=2 考虑自旋,电子分为α,β电子计算,考虑磁性
- ISPIN=2时,可指定体系的初始磁矩MAGMOM
输入格式:
MAGMOM=原子种类1*自旋磁矩 原子种类2*自旋磁矩 ...
MAGMOM=2*1
MAGMOM=2 2
还可以更大一点
MAGMOM=5 5(2*5)
注意 两个数字之间不能有空格2*5 2 * 5后面是不对的
举例子:
对于Ni C H O 这四种元素组成的物质 MAGMOM可以写成
MAGMOM=56*1 17*0 20*0 6*0
对于简单体系来说,MAGMOM可以采用默认值;
MAGMOM设置的时候,初始值不要求与实验值完全一致,一般取大些(1.5倍)比较好
POSCAR
POSCAR文件主要是存放待计算结构的信息 格式如下
AlN bulk (Title) 首行注释
1.0 缩放系数
3.11 0.00 0.00 第一个平移矢量a方向
-1.56 2.69 0.00 第二个平移矢量b方向
0.00 0.00 4.98 第三个平移矢量c方向
Al N 原子种类
2 2 单胞内原子数目
Selective dynamics [可选]有对构型(原子坐标)进行部分优化,没有,则全优化
Direct Direct分数坐标,Car实际坐标单位为埃
0.667 0.333 0.000 T T T T表示对此方向优化,F表示对此方向不优化
0.333 0.667 0.500 T T T
0.667 0.333 0.382 T T T
0.333 0.667 0.882 F F F
POTCAR
主要是演示文件 来任务了 今天先写到这里
CONTCAR
记录优化后的原子坐标位置,一般后面还会有一堆,都是零,如果跑分子动力学就不是零 代表速度, 不然其他的基本都是零。
除了记录优化后的原子的坐标位置,还可以在断电之后或者意外中断后续算:
续算分三种情况:
A) 第一个离子步没有算完,任务就挂掉了。这种情况,CONTCAR是不会更新的,我们再次用原来的输入文件提交一次就行了。
B)我们的计算已经完成了大于或者等于1的离子步,但小于INCAR中设置的NSW的数值。这个时候CONTCAR的内容已经是离任务死掉最近的结构了。我们只需要将其复制成POSCAR,然后再次提交任务即可。具体操作如下:
$ mv POSCAR POSCAR_0
$ mv OUTCAR OUTCAR_0
$ cp CONTCAR POSCAR
C)我们的计算达到的INCAR中所设置的NSW的数值。比如设置的NSW = 1000,实际上跑了1000步,任务停下来了,也就是所谓的结构优化没有收敛。这种情况我们需要做的又有2个步骤:
-
首先,要检查CONTCAR中的结构是不是正确的,如果结构跑乱了,体系中原子乱飞,有很大可能会导致不收敛的情况。如果是结构乱了,我们就要找原因去解决。主要还是在以下三个方向下功夫:
- 初始结构是否合理
- POSCAR中的元素顺序与POTCAR中的是不是一致
- 是不是用的gamma点,然后把体系放开了。
-
如果前面检查的结构没问题,这种情况,可能是因为你设置的NSW值太小导致的,或者体系是在是太难收敛,比如过渡态优化的情况。那么我们就需要继续算了。此时,为了保证计算的可重复性,我们必须要将上一步的计算保存记录下来。
OSZICAR
计算一个体系,我们有2个优化过程:
1.电子结构的优化: 可以理解为对某一固定的几何结构,迭代求解薛定谔方程来获得体系能量极小值的一个过程。这个迭代过程,每一次迭代求解都可以认为是电子结构的一个优化。(通常被大伙称为:电子步)
2.几何结构的优化:可以理解为在电子结构优化的结果上,获取原子的受力情况,然后根据受力情况,调节原子的位置,再进行电子结构优化,获取新的受力情况,然后再调节原子位置,一直重复这样的过程,直至找到体系势能面上一个极小值的过程。(通常被大伙称为:离子步)
OSZICAR是用来记录优化过程一些信息的文件。这里的优化过程既包括电子结构,又包括几何结构。
OSZICAR 张这样子:
N E dE d eps ncg rms rms(c)
DAV: 1 0.324969965196E+02 0.32497E+02 -0.10270E+03 48 0.977E+01
DAV: 2 0.501749892771E+00 -0.31995E+02 -0.31995E+02 72 0.202E+01
DAV: 3 -0.182605770767E-01 -0.52001E+00 -0.50521E+00 48 0.521E+00
DAV: 4 -0.203547758465E-01 -0.20942E-02 -0.20860E-02 96 0.333E-01
DAV: 5 -0.203547873947E-01 -0.11548E-07 -0.11210E-07 48 0.844E-04 0.307E-01
DAV: 6 -0.213726161828E-01 -0.10178E-02 -0.17884E-03 48 0.111E-01 0.155E-01
DAV: 7 -0.214708381542E-01 -0.98222E-04 -0.23522E-04 48 0.459E-02
1 F= -.21470838E-01 E0= -.13757722E-01 d E =-.154262E-01
第一行中各项的含义:(没汉语解释的,大师兄也翻译不出来)
1) N 代表电子结构的迭代步数,通常被大家称为电子步。
2) E 代表当前电子步的体系能量;
3) dE当前电子步和上一步体系能量的差值;
4) d eps the change in the band structure energy;
5)ncg the number of evaluations of the Hamiltonian acting onto a wavefunction;
6) rms the norm of the residuum of the trialwavefunctions (i.e. their approximate error)
7) rms (c) the difference between input and output charge density.
dev 为某种电子迭代自洽算法(还有RMM CG等)
最后一行:
1) F前面的 1 代表几何结构优化的次数(也称为离子步的步数)。
2) F = 是体系的总能量, 与OUTCAR中 free energy TOTEN 后面的值相等;
3) E0 后面的能量对应OUTCAR中 energy (sigma->0)后面的能量。
通过OSZICAR获取体系的能量,也就是E0后面的那一项。很多人在使用VASP的时候,不知道该选择哪个能量,这里大师兄就告诉你:选择E0后面的即可。不管你有什么疑问,不管别人怎么跟你争论,都不要管,先老老实实记住:我们选E0后面的这个能量。随着你的学习,很多疑问自己就解开了。命令使用方式:
$ grep E0 OSZICAR
1 F= -.21470838E-01 E0= -.13757722E-01 d E =-.154262E-01
在OUTCAR中与E0对应(相同)的是energy(sigma->0) = 后面的那个.
提取能量的命令:
grep without OUTCAR | tail -n 1
grep ' without' OUTCAR | tail -n 1 # bigbro常用的是这个
grep sigma OUTCAR | tail -n 1
OUTCAR中的Entropy表示的不是传统意义上的熵
费米能 E-fermi
电子占据情况:在OUTCAR里。
判断收敛情况:
$ grep reached OUTCAR
------------------------ aborting loop because EDIFF is reached ----------------------------------------
------------------------ aborting loop because EDIFF is reached ----------------------------------------
------------------------ aborting loop because EDIFF is reached ----------------------------------------
reached required accuracy - stopping structural energy minimisation
4 $ grep reached OUTCAR |tail -n 1 reached required accuracy - stopping structural energy minimisation
or
grep 'reached required accuracy' OUTCAR
可以写在.bashrc文件中
$ alias gr="grep 'reach required accuracy' OUTCAR"
$ source .bashrc
$ . .bashrc
如果发现没有收敛 查看OSZICAR 看电子步和离子步的收敛情况 如果最后一步电子步是六十 那么很可能是电子步达到了NELM默认的60后自动停止计算
这时候需要考虑初始结构的合理性以及增加NELM继续算下去
电子步(SCF): EDIFF <====> NELM
离子步(结构优化):EDIFFG <====> NSW
一般来讲,可是增加到NELM=100 难收敛的可以到200,同时也要注意能量变化 能量变化吉大也可能是结构的问题
如果跑了很久每个离子步中的电子部都不收敛 可以尝试换个电子步迭代的算法 这种情况ALGO=ALL 结合NELM=200可以解决大多数问题(比较耗时)